Penyakit genetik
definisi
Penyakit genetik atau penyakit keturunan adalah penyakit yang disebabkan oleh satu atau lebih gen orang yang bersangkutan. Di sini DNA bertindak sebagai pemicu langsung penyakit. Untuk kebanyakan penyakit genetik, lokasi gen penyebab diketahui. Jika diduga ada penyakit genetik, diagnosis terkait dapat dibuat melalui pemeriksaan genetik.
Di sisi lain, ada pula sejumlah penyakit yang kemunculannya memiliki pengaruh genetik atau sedang dibahas, seperti diabetes mellitus (“diabetes”), osteoporosis, atau depresi. Ini yang disebut disposisi, yaitu kemungkinan peningkatan penyakit tertentu. Disposisi harus dibedakan dari penyakit keturunan.

Ini adalah penyakit keturunan yang umum
Secara absolut, penyakit keturunan tidak umum, tetapi penyakit keturunan yang tercantum di sini sering terjadi dibandingkan dengan penyakit penyebab genetik lainnya.
-
Sindrom Marfan
-
Anemia sel sabit
-
Hemofilia (hemofilia A atau B)
-
Faktor V Leiden mutasi dan menghasilkan resistensi APC
-
Kelemahan Merah Hijau
-
Defisiensi glukosa-6-fosfat dehidrogenase (defisiensi G6PD)
-
Polydactyly ("banyak jari", juga mungkin sebagai gejala penyakit lain)
-
Trisomi 21 (sindrom Down)
-
Chorea huntington
penyebab
Penyakit keturunan sangat beragam dalam penampilannya. Mereka pada dasarnya hanya memiliki satu kesamaan: Penyebab masing-masing terletak pada DNA, yaitu pada materi genetik orang yang bersangkutan. Berbagai perubahan dapat terjadi di sini, seperti mutasi (pertukaran informasi DNA) atau penghapusan (kekurangan materi genetik tertentu).
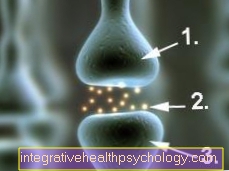
Sejumlah besar informasi dikodekan dalam materi genetik, seperti "cetak biru" untuk berbagai komponen yang penting untuk berfungsinya sel tubuh. Ini bisa berupa enzim, saluran elektrolit atau zat pembawa pesan, misalnya. Unsur-unsur terkecil ini kemudian salah dibaca atau tidak dibaca sama sekali dari DNA, yang kemudian hilang dalam sistem canggih tubuh. Informasi genetik yang salah atau hilang menyebabkan kerusakan tertentu dalam tubuh. Ini kemudian menyebabkan gejala sesuai dengan sistem fungsional di mana satu elemen sekarang hilang.
Cari tahu semua tentang topik tersebut di sini: Tes genetik.
Beginilah cara penyakit keturunan diturunkan
Setiap penyakit keturunan diwariskan baik secara monogenetis maupun poligenetik: Artinya ada satu atau lebih lokasi genetik yang harus diubah untuk mengarah pada suatu penyakit.
Lebih jauh, ciri-ciri genetik selalu dapat diwariskan secara dominan atau resesif: Resesif berarti bahwa pasti ada kecenderungan untuk penyakit keturunan tertentu ini pada gen ayah dan ibu. Dalam kasus warisan dominan, satu perubahan (yaitu satu orang tua) sudah cukup untuk memicu penyakit. Oleh karena itu, pada penyakit yang diturunkan secara dominan, orang-orang yang menjadi karier juga akan jatuh sakit - sementara dengan pewarisan resesif biasanya bahkan tidak diketahui adanya kecenderungan genetik yang sesuai.
Ada juga penyakit yang diturunkan melalui kromosom seks, seperti hemofilia atau kebutaan merah-hijau. Fasilitas untuk ini biasanya ada pada kromosom X, karena kromosom Y secara keseluruhan sangat kecil dan umumnya hanya dapat menyimpan sedikit informasi genetik. Oleh karena itu, seseorang berbicara tentang penyakit keturunan terkait-X. Ini biasanya mempengaruhi lebih banyak pria daripada wanita, karena wanita dapat mengkompensasi informasi yang salah pada kromosom X dengan yang kedua.
Bagaimana tepatnya suatu penyakit genetik diturunkan biasanya mudah untuk diteliti jika Anda tertarik.
Tes sebelum lahir
Pada prinsipnya materi genetik anak sudah dapat diperiksa di dalam kandungan untuk semua penyakit keturunan yang diketahui lokasi genetik penyebabnya. Namun, analisis genetik memakan waktu, jadi biasanya hanya lokasi gen yang dicurigai yang dianalisis - untuk ini, pada gilirannya, harus ada kecurigaan yang dapat dibenarkan tentang penyakit genetik.
Untuk pemeriksaan semacam itu, materi genetik kemudian dapat diambil dari cairan ketuban atau plasenta dan digunakan untuk analisis.
Namun, harus selalu diingat bahwa setiap diagnosis invasif juga berisiko terhadap kehidupan bayi yang belum lahir. Karena itu, tusukan seperti itu harus ditimbang secara individual dalam setiap kasus.
Ada juga pengukuran yang dapat mengindikasikan penyakit genetik, seperti pengukuran transparansi nukal sebagai tanda trisomi 21. Metode tersebut tidak berbahaya untuk janin, tetapi tidak dapat memberikan kepastian mutlak bahwa ada penyakit genetik. Jadi di sini juga, operasi harus dipertimbangkan dengan cermat.
Trisomi 21
Penyebab trisomi 21 adalah kromosom 21, yang tidak muncul dua kali tetapi tiga kali pada orang yang terkena. Varian DNA ini dibuat ketika kromosom didistribusikan di sel germinal induk, yaitu sel sperma atau sel telur. Oleh karena itu, ini adalah "kesalahan distribusi" dan bukan perubahan materi genetik yang sebenarnya. Ini menjelaskan mengapa trisomi 21 dapat terjadi secara spontan di setiap keluarga dan mengapa kemungkinan memiliki anak dengan sindrom Down sama di semua keluarga. Sebenarnya, trisomi 21 - seperti trisomi lainnya - tidak boleh dianggap sebagai penyakit keturunan dalam arti sebenarnya. Namun demikian, trisomi 21 adalah penyakit terkait DNA yang paling umum pada bayi baru lahir.
Ciri-ciri perubahan set kromosom pada sindrom Down sudah dapat dilihat pada janin dalam kandungan: Keterlambatan pertumbuhan dan cacat dapat menyebabkan, antara lain, tengkorak yang terlalu kecil, tulang paha dan lengan atas yang pendek, serta kelainan jantung. Cairan ketuban dalam jumlah besar juga dapat menjadi indikasi trisomi 21, karena bayi yang belum lahir minum atau menelan sedikit cairan ketuban. Namun, tidak satu pun dari ciri-ciri ini yang merupakan tanda pasti dari sindrom Down!
Selain tanda-tanda retardasi pertumbuhan yang disebutkan, anak-anak dengan sindroma Down seringkali juga menunjukkan perkembangan yang terhambat, misalnya dalam bidang bahasa dan keterampilan motorik. Orang yang terkena sindrom Down sering kali menunjukkan keterampilan sosial yang luar biasa, sementara kecerdasannya sering kali berada di bawah rata-rata. Namun, orang yang terkena dampak sangat berbeda dalam karakteristik ini, dan tidak jarang mereka lulus dari sekolah setelah mendapat dukungan yang baik.
Di kemudian hari, penderita trisomi 21 memiliki risiko lebih tinggi untuk didiagnosis dengan penyakit tertentu. Ini termasuk penyakit Alzheimer, epilepsi dan kanker, terutama leukemia. Namun demikian, angka harapan hidup penderita Down Syndrome terus meningkat: Sementara itu, orang yang terkena sering mencapai usia 60 atau 70 tahun.
Anda dapat menemukan informasi lebih lanjut di situs web kami Sindrom Down
Defisiensi antitripsin alfa-1
Kekurangan antitripsin alfa-1 dapat terjadi dalam berbagai bentuk dan bentuk, tergantung pada karakteristik genetik orang yang terkena. Ini berarti bahwa tidak setiap defisiensi antitripsin alfa-1 menyebabkan gejala. Berikut ini, hanya jenis yang secara klinis mencolok (PiZZ) dari penyakit yang ditentukan secara genetik ini yang akan dibahas.
Cacat enzim yang ada pada penyakit ini menyebabkan kerusakan dan pembentukan kembali blok bangunan di jaringan organ pada orang yang terkena. Selain itu, protein yang rusak disaring dari darah oleh hati dan menumpuk di sana. Hal ini dapat menyebabkan peradangan hati (hepatitis), sirosis atau kanker hati. Saluran udara di paru-paru menjadi tidak stabil karena kurangnya jaringan yang stabil dan lebih cepat runtuh: Gambaran klinis PPOK (penyakit paru obstruktif kronik) berkembang. Gambaran klinis ini seringkali merupakan gejala pertama dari defisiensi antitripsin alfa-1, jadi setiap orang dengan PPOK pada usia yang lebih muda harus diperiksa untuk defisiensi antitripsin alfa-1.
Jika penyakitnya telah berlangsung lama, paru-paru bisa membengkak karena udara yang Anda hirup tidak dapat dihembuskan dengan baik melalui saluran udara yang tidak stabil dan menumpuk di paru-paru. Sebagai terapi, selain secara konsisten menghindari merokok dan vaksinasi rutin untuk mencegah penyakit pernapasan, tindakan pengobatan juga harus dilakukan: Alfa-1-antitripsin yang hilang dapat diberikan secara intravena untuk meringankan gejala sejauh mungkin dan menghentikan perjalanan penyakit.
Anda dapat menemukan informasi lebih lanjut di situs web kami Defisiensi antitripsin alfa-1
hemofilia
Kelompok hemofilia juga dikenal dalam bahasa sehari-hari sebagai "hemofilia", karena istilah ini menggambarkan gejala utama penyakit keturunan ini dengan sangat tepat: orang yang terkena mengalami pendarahan lebih lama dan, tergantung pada tingkat keparahan penyakitnya, lebih sering daripada tidak terkena.
Pendarahan biasanya dihentikan oleh apa yang dikenal sebagai kaskade koagulasi, jalur pensinyalan endogen yang mencegah kehilangan darah berlebihan. Dalam sistem koagulasi ini, 13 faktor berperan, yang mengaktifkan satu sama lain satu demi satu. Ini bisa dibayangkan sebagai rangkaian kartu domino: jika Anda menabrak satu batu (faktor koagulasi), batu berikutnya mengaktifkan, dan seterusnya. Di ujung jalur sinyal atau domino ini terjadi pembekuan darah. Dengan hemofilia, faktor tertentu hilang - tergantung pada subtipe spesifik penyakitnya: reaksi berantai terputus di sini.
Terapi penyakit dapat dilakukan dengan menentukan faktor yang hilang dan menambahkannya dari luar. Oleh karena itu, orang yang terkena dampak harus secara teratur menyuntikkan diri mereka sendiri dengan sediaan dengan faktor koagulasi ini sehingga sisa reaksi berantai dapat berlangsung.
Anda dapat menemukan informasi lebih lanjut di situs web kami Penyakit darah
Fibrosis kistik
Dalam penyakit genetik fibrosis kistik - juga dikenal sebagai fibrosis kistik - ada produksi saluran ion yang salah, lebih tepatnya saluran klorida. Akibatnya, komposisi sekresi tubuh (misalnya keringat, sekresi dari saluran pernapasan, dan pankreas) dari orang yang terkena berubah: Karena kekurangan klorida berarti lebih sedikit air yang ditarik ke dalam saluran kelenjar masing-masing, sekresi relatif kental.
Akibatnya, gejala biasanya berkembang di saluran pencernaan, karena sekresi enzim pencernaan tidak dapat mengalir dengan baik dari pankreas ke usus dan dengan demikian merusak pankreas itu sendiri. Selain itu, gangguan pencernaan seperti tinja berlemak, diare, dan berat badan rendah sering terjadi.
Kelompok besar kedua gejala biasanya berkembang di paru-paru: Karena lendir yang secara alami muncul di paru-paru lebih kental daripada pada orang sehat, lebih sulit untuk mengeluarkannya dari silia. Ini dapat menyebabkan batuk kronis dan penyumbatan pada bronkus (bronkiektasis). Jumlah sekresi paru yang lebih besar juga menyediakan lingkungan yang baik untuk pertumbuhan bakteri, yang mengakibatkan seringnya infeksi saluran pernapasan dan pneumonia.
Fibrosis kistik diobati sesuai gejalanya dengan ekspektoran, enzim pencernaan, dan antibiotik untuk infeksi.
Anda dapat menemukan lebih banyak tentang ini di situs web kami Fibrosis kistik
Faktor V Leiden dan Resistensi APC
Mutasi faktor V Leiden melibatkan perubahan informasi genetik yang dapat menyebabkan peningkatan pembekuan darah. Alasannya adalah faktor V dalam tubuh yang disebut kaskade koagulasi: jalur sinyal ini memastikan bahwa jika terjadi cedera, luka ditutup oleh "protein perekat" (fibrin) tubuh sendiri. Ada 13 faktor dalam jalur pensinyalan ini, yang diberi nama dengan angka romawi (artinya “Faktor 5 penderitaan”!). Faktor V memiliki efek menguntungkan pada pembentukan sumbat fibrin, tetapi juga dapat dihambat oleh apa yang disebut protein C aktif (disingkat APC). Ini memainkan peran penting dalam mengatur jalur pensinyalan ini dan dalam mencegah pembekuan darah yang berlebihan.
Faktor V yang bermutasi ada pada individu yang terkena tetapi tidak merespons APC. Tubuh kekurangan "perangkat keamanan" penting pada saat ini untuk mencegah pembekuan darah tanpa alasan, yang bahkan dapat menyumbat pembuluh dan dengan demikian menyebabkan gangguan peredaran darah.
Secara statistik, orang yang dipengaruhi oleh mutasi faktor V Leiden lebih mungkin menderita peristiwa trombotik (yaitu trombosis atau emboli paru), bahkan tanpa riwayat faktor risiko yang khas. Dalam istilah teknis, seseorang juga berbicara tentang "trombofilia", yaitu kecenderungan untuk menggumpal.
Anda dapat menemukan lebih banyak tentang ini di situs web kami Faktor V Leiden
Penyakit Gaucher
Pada penyakit Gaucher, perubahan informasi DNA menyebabkan kerusakan pada enzim yang terlibat dalam metabolisme lipid, lebih tepatnya glukoserebrosidase: Ini membantu memecah komponen sel tua. Jika terjadi kerusakan, dapat terjadi penurunan fungsionalitas atau bahkan hilangnya fungsionalitas, dan karenanya gejala muncul di masa kanak-kanak atau dewasa muda.
Gejala penyakit Gaucher sebagian besar disebabkan oleh pembesaran hati dan limpa, yang pertumbuhannya dicoba oleh tubuh untuk mengimbangi kekurangan enzim. Hal ini meningkatkan pemecahan semua komponen darah, yang dapat dikenali dalam hitung darah dan digunakan sebagai indikator diagnostik bersama dengan pembesaran hati dan limpa.
Enzim glukoserebrosidase yang hilang dapat digunakan sebagai terapi sebagai obat. Prognosis dan perjalanan penyakit Gaucher sangat bergantung pada tingkat keparahan hilangnya fungsi enzim.
Untuk informasi lebih lanjut, baca di sini: Penyakit Gaucher.
Penyakit Osler
Penyakit Osler adalah penyakit keturunan yang ditandai dengan vasodilatasi yang kuat. Pada prinsipnya, perluasan pembuluh darah ini bisa terjadi di mana saja, baik di kulit maupun di organ dalam. Dinding pembuluh yang membesar relatif tipis dan mudah robek. Akibatnya, daerah yang terkena bencana cepat berdarah.
Vasodilatasi sering terjadi pada wajah dan selaput lendir hidung, sehingga orang yang terkena biasanya mengeluhkan sering mimisan dan perdarahan bercak kecil di wajah.
Jika penyakit Osler dicurigai, diagnosa yang tepat harus dilakukan, karena vasodilatasi juga dapat terjadi pada organ atau organ vital dengan suplai darah yang baik, seperti paru-paru, otak atau hati, di mana perdarahan dari pembuluh yang pecah berbahaya.
Anda dapat menemukan lebih banyak tentang topik ini di situs web kami Penyakit Osler
Penyakit Recklinghausen
Neurofibromatosis tipe 1 - atau penyakit Recklinghausen - adalah penyakit genetik di mana mereka yang terkena sering mengembangkan tumor pada sel-sel penutup saraf. Tumor yang berkembang bisa jinak dan ganas dan muncul di usia muda.
Tumor tipikal, bagaimanapun, adalah neurofibroma jinak: Ini terdiri dari sel-sel yang melapisi dan mengisolasi saraf seperti kabel listrik, serta jaringan ikat di sekitarnya. Mereka jinak, yaitu tumor yang tidak menyebar dan tumbuh perlahan.
Namun, pembedahan untuk menghilangkan neurofibroma bisa jadi sulit, karena sering kali neurofibroma melekat erat pada saraf dan saraf yang terkait kemudian harus diangkat. Namun demikian, ini adalah satu-satunya pilihan pengobatan untuk neurofibroma simptomatik, karena terapi kausal untuk penyakit keturunan ini tidak memungkinkan.
Anda dapat menemukan lebih banyak tentang topik ini di situs web kami Neurofibromatosis tipe 1
Distrofi otot
Istilah distrofi otot menggambarkan sekelompok penyakit keturunan di mana komponen otot tertentu tidak dapat atau tidak dapat dirakit dengan benar oleh sel-sel tubuh. Akibatnya, orang yang terkena biasanya mengalami kelemahan otot sejak masa kanak-kanak dan remaja, dan ini dapat mengakibatkan hilangnya massa otot, pembatasan gerakan, dan bahkan cacat fisik.
Jika dicurigai adanya distrofi otot, nilai darah harus ditentukan terlebih dahulu. Jika nilainya sesuai dengan diagnosis yang dicurigai, biopsi otot masih dapat dilakukan: Sampel jaringan kecil diambil dari otot, yang kemudian diperiksa secara mikroskopis untuk mengetahui adanya kerusakan sel. Pemeriksaan genetik juga memungkinkan untuk menegakkan diagnosis, karena lokasi genetik yang sesuai biasanya dikenal untuk berbagai bentuk distrofi otot dan harus diubah. Terapi kausal untuk distrofi otot tidak diketahui.
Anda dapat menemukan lebih banyak tentang topik ini di situs web kami Distrofi otot
Xeroderma pigmentosum
Xeroderma pigmentosum adalah penyakit keturunan langka di mana enzim tertentu di kulit orang yang terkena tidak bekerja. Enzim-enzim ini biasanya menjaga perbaikan dalam DNA, yang dapat rusak oleh sinar matahari atau sinar UVB yang terkandung. Kerusakan UVB dapat menyebabkan kanker kulit pada orang yang terkena dampak serta pada semua orang lainnya, tetapi dengan Xeroderma Pigmentosum prosesnya dipercepat oleh kurangnya mekanisme perbaikan. Akibatnya, orang yang terkena dampak mengembangkan bentuk kanker kulit yang parah di masa kanak-kanak dan remaja dan setelah terpapar sinar matahari sebentar.
Terapi kausal belum memungkinkan. Orang-orang yang terkena dampak harus menghindari sinar matahari seumur hidup, itulah sebabnya julukan "anak-anak bulan purnama" telah ditetapkan untuk mereka yang terkena dampak (terkadang sangat muda). Selain itu, orang-orang ini harus diawasi oleh dokter kulit untuk melakukan skrining kanker kulit secara teratur agar dapat segera menghilangkan kanker kulit yang baru berkembang. Jika langkah-langkah ini diikuti dengan ketat, harapan hidup seseorang dengan xeroderma pigmentosum hampir sama dengan orang yang tidak terkena.
Anda dapat menemukan lebih banyak tentang penyakit ini di situs web kami Xeroderma pigmentosum
Sindrom Lynch
Sindrom Lynch adalah perubahan DNA yang menyebabkan kerusakan enzim pada sel tubuh.Pada orang yang terkena, mekanisme tertentu oleh karena itu rusak, yang seharusnya melindungi sel dari degenerasi, yaitu pertumbuhan yang tidak terkendali - oleh karena itu orang dengan sindrom Lynch memiliki risiko sangat tinggi terkena kanker.
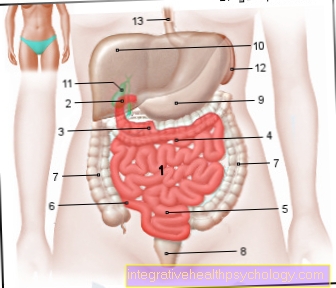
Kanker usus besar sering terjadi karena sel-sel secara alami sering membelah di sini dan kesalahan dalam pemrograman pertumbuhan dan kematian sel menjadi lebih cepat terlihat. Orang yang terkena sering mengembangkan tumor di usus besar pada usia yang sangat muda, yaitu sebelum usia 50, yang kemudian disebut HNPCC (kanker usus besar non-polip herediter). Namun, tidak semua orang yang memiliki susunan genetik sindrom Lynch akan mengembangkan kanker usus besar. Di sisi lain, organ lain juga dapat mengembangkan tumor, karena kecenderungan genetik yang mendukung perkembangan tumor terdapat di semua sel tubuh. Oleh karena itu, pemeriksaan rutin dan pemeriksaan pencegahan diperlukan bagi mereka yang terkena sindrom Lynch untuk mengobati tumor yang berkembang pada tahap awal secara memadai.
Anda dapat menemukan lebih banyak tentang topik ini di situs web kami Sindrom Lynch